
On poursuit sur l’exploration des démences… Attention, ces billets sont denses, et évidemment pas rigolos… C’est vraiment pour ceux que ça intéresse ! ^^
Les critères diagnostiques cités dans le premier billet sur les démences permettent de retenir un diagnostic de démence. La démence est un syndrome clinique causé par la neuro-dégénérescence, qui implique un déclin progressif des capacités cognitives et de la capacité de mener une vie autonome [21]. Nous aborderons ici les principaux types de démences, repris dans la classification de la CIM-10 : la démence d’Alzheimer, la démence vasculaire, la démence à corps de Lewy, la démence mixte, la dégénérescence lobaire fronto-temporale, la démence secondaire à l’abus de substances psychoactives et les troubles cognitifs liés à d’autres affections. Nous détaillerons uniquement les différents critères diagnostiques utilisées en 2018 pour la maladie d’Alzheimer.
Commençons par un petit résumé visuel…
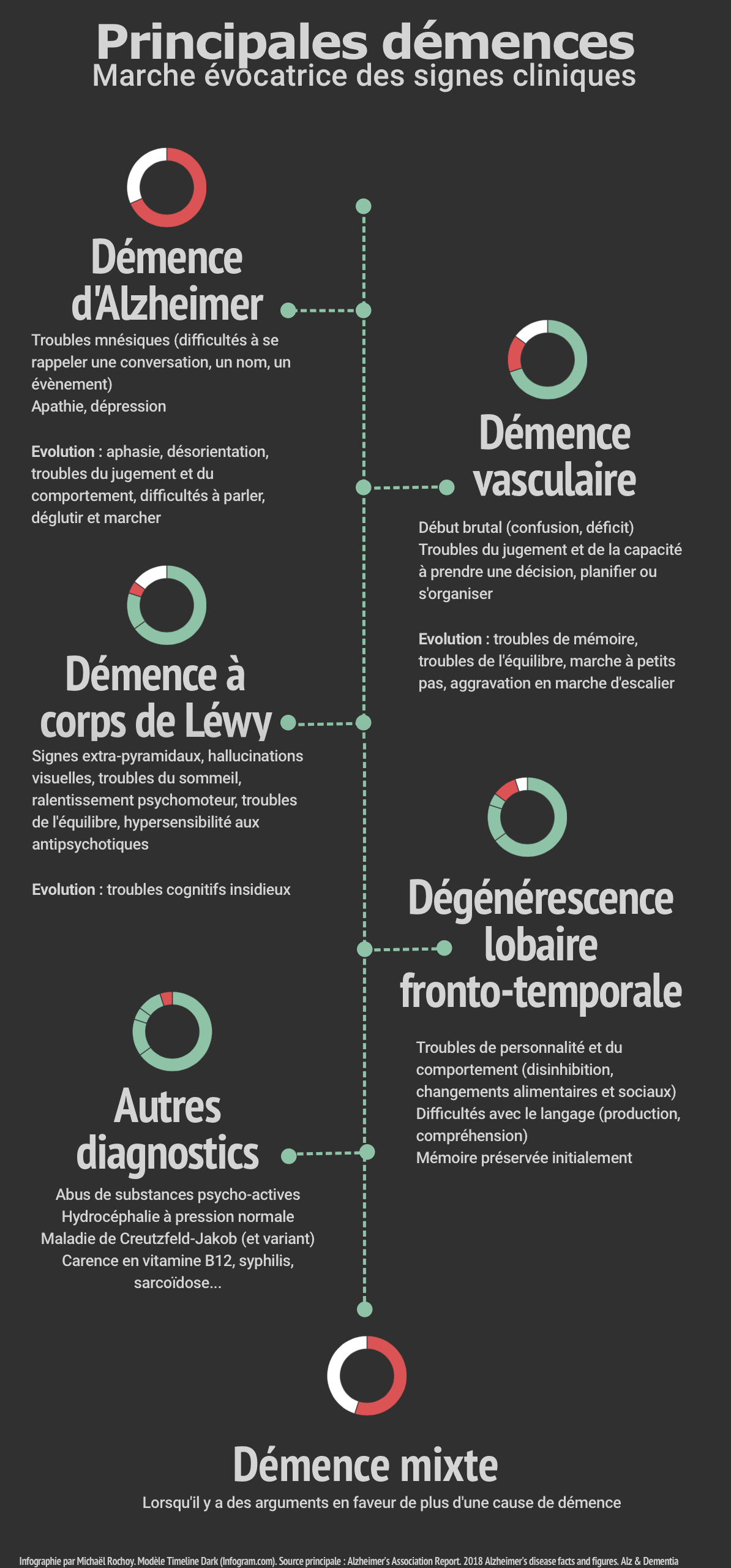
1.1.1 Maladie et démence d’Alzheimer
La démence d’Alzheimer représente 60 à 70 % des démences [22,23]. Il s’agit d’une des principales causes de morbi-mortalité dans la population âgée. Elle a été décrite en 1906 par le docteur Alois Alzheimer (Annexe 1). Il existe une période pré-symptomatique d’une quinzaine d’années entre les modifications biochimiques cérébrales et le développement d’une démence d’Alzheimer : cette phase pré-démentielle est également appelée « maladie » d’Alzheimer [24,25].
La physiopathologie de la maladie d’Alzheimer est complexe et encore mal connue en 2018. Les principales hypothèses sont :
- la surproduction de peptides amyloïdes (mutations d’APP, de préseniline 1 ou 2 du complexe gamma-secrétase),
- le défaut d’élimination des peptides amyloïdes (l’allèle e4 du gène de l’apoprotéine E (ApoEe4) diminuerait la clairance des peptides [26])
- la phosphorylation de protéines tau, transmises entre les neurones [27],
La plainte initiale concerne les troubles de mémoire. Ils sont suivis d’une perte progressive d’autres fonctions cognitives (fonctions exécutives, attention, langage, cognition et jugement social, vitesse psychomotrice, capacités visuo-perceptuelles ou visuo-spatiales) et de troubles mentaux et du comportement (humeur dépressive, apathie, symptômes psychotiques, irritabilité, agressivité, confusion, anomalies de la mobilité et convulsions) [17].
Pour poser le diagnostic, des critères précis de la maladie d’Alzheimer sont définis par la CIM-10, le DSM-5 ainsi que par le National Institute of Neurological and Communicative Diseases and Stroke-Alzheimer’s Disease and Related Disorders Association(NINCDS-ADRDA) [28,29]. En 1999, Varma et al. ont montré que ces derniers critères ne permettent pas de différencier la démence d’Alzheimer de la démence frontotemporale [30]. Les connaissances sur la physiopathologie de la maladie d’Alzheimer évoluant, une révision des critères du NINCDS-ADRD a été proposée en 2007 [29].
1.1.1.1 Critères diagnostiques de la NINCDS-ADRA (1984)
Dans les critères NINCDS-ADRA, la maladie d’Alzheimer peut être probable, possible ou certaine.
Les critères pour une maladie d’Alzheimer probable sont :
- syndrome démentiel établi sur des bases cliniques et documenté par le Mini-Mental State Examination, le Blessed Dementia Scale ou tout autre test équivalent et confirmé par des épreuves neuropsychologiques ;
- et déficits d’au moins deux fonctions cognitives ;
- et altérations progressives de la mémoire et des autres fonctions cognitives ;
- et absence de trouble de conscience ;
- et survenue entre 40 et 90 ans le plus souvent au-delà de 65 ans ;
- et absence de désordres systémiques ou d’une autre maladie cérébrale pouvant rendre compte des déficits mnésiques et cognitifs progressifs.
Ce diagnostic est renforcé par :
- la détérioration progressive des fonctions telles que le langage (aphasie), les habiletés motrices (apraxie) et perceptives (agnosie) ;
- ou la perturbation des activités de vie quotidienne et la présence de troubles du comportement ;
- ou une histoire familiale de troubles similaires surtout si confirmés histologiquement ;
- ou le résultat aux examens standards suivants : normalité du liquide céphalo-rachidien ; EEG normal ou siège de perturbations non-spécifiques comme la présence d’ondes lentes ; présence d’atrophie cérébrale d’aggravation progressive.
D’autres caractéristiques cliniques sont compatibles après exclusion d’autres causes :
- périodes de plateaux au cours de l’évolution ;
- ou présence de symptômes tels que dépression, insomnie, incontinence, idées délirantes, illusions, hallucinations, réactions de catastrophes, désordres sexuels et perte de poids. anomalies neurologiques sont possibles surtout aux stades évolués de la maladie, notamment des signes moteurs tels qu’une hypertonie, des myoclonies ou des troubles de la marche ;
- ou crises comitiales aux stades tardifs ;
- ou scanner cérébral normal pour l’âge.
A l’inverse, des signes rendent le diagnostic de « maladie d’Alzheimer probable » incertain ou improbable :
- début brutal ;
- ou déficit neurologique focal tel qu’hémiparésie, hypoesthésie, déficit du champ visuel, incoordination motrice à un stade précoce ;
- ou crises convulsives ou troubles de la marche en tout début de maladie.
Les critères pour une maladie d’Alzheimer possibles sont :
- syndrome démentiel, en l’absence d’autre désordre neurologique, psychiatrique ou systémique susceptible de causer une démence, et en présence de variante dans la survenue, la présentation ou le cours de la maladie ;
- ou présence d’une seconde maladie systémique ou cérébrale susceptible de produire un syndrome démentiel mais qui n’est pas considéré comme la cause de cette démence ;
- ou, en recherche clinique, quand un déficit cognitif sévère progressif est identifié en l’absence d’autre cause identifiable.
Enfin, la maladie d’Alzheimer est certaine en présence :
- des critères cliniques de maladie d’Alzheimer probable ;
- et d’une preuve histologique apportée par la biopsie ou l’autopsie.
1.1.1.2 Proposition de révision des critères diagnostiques de la NINCDS-ADRA (2007)
La proposition de révision retient le diagnostic de maladie d’Alzheimer probable en présence d’un critère majeur, et d’un ou plusieurs des 4 critères secondaires.
Le critère majeur est un trouble de mémoire épisodique initial, constitué par :
- des troubles de mémoire fonctionnels progressifs rapportés par le patient ou l’entourage depuis au moins six mois ;
- et la mise en évidence d’un trouble de mémoire épisodique significatif dans les tests avec un déficit de rappel non significativement amélioré ou non normalisé en situation d’indiçage ou de reconnaissance, alors que l’encodage initial de l’information a été contrôlé.
Les 4 critères secondaires sont :
- une atrophie des structures temporales internes : atrophie hippocampique, entorhinale ou amygdalienne mise en évidence en IRM par échelle visuelle qualitative ou par volumétrie quantitative, en référence à des sujets témoins de même âge ;
- une modification du taux de biomarqueurs dans le LCR : diminution des taux d’Aβ1-42et/ou augmentation de la concentration totale de protéine Tau ou de phospho-Tau et/ou modification de tout autre marqueur validé dans le futur ;
- un profil spécifique à la tomographie par émission monophotonique (TEMP) ou à la tomographie par émission de positons (TEP) : diminution du métabolisme du glucose dans les régions temporo-pariétales bilatérales et/ou toute autre anomalie de distribution de ligand validée dans l’avenir ;
- une mutation autosomale dominante dans la famille directe.
Les critères d’exclusion peuvent être liés au mode d’installation ou à la présentation clinique :
- début brutal ;
- survenue précoce de troubles de la marche, de crises comitiales, de troubles comportementaux ;
- déficit neurologique focal : hémiparésie, troubles sensoriels, déficit du champ visuel ;
- signes extrapyramidaux précoces ;
- conditions médicales pouvant rendre compte, à elles seules, des troubles de mémoire ou cognitifs : autres démences, dépression majeure, pathologie cérébrovasculaire, troubles métaboliques ou toxiques ;
- anomalies IRM en FLAIR ou en T2 dans la région temporale interne, évoquant une atteinte infectieuse ou vasculaire.
La maladie d’Alzheimer est considérée comme définie en présence :
- du critère majeur ;
- et d’une preuve histologique (biopsie corticale ou autopsie) ou génétique (mutation génique sur le chromosome 1, 14 ou 21)
1.1.1.3 Critères diagnostiques du DSM-5 (2013)
Le diagnostic de maladie d’Alzheimer repose sur les 3 critères suivants :
- critères de l’atteinte neurocognitive légère ou sévère ;
- et installation insidieuse des symptômes et déclin graduel dans un ou plusieurs domaines cognitifs ;
- et pas d’autre atteinte à la santé mentale ou physique pouvant expliquer les déficits observés.
Le DSM-5 distingue la forme « sévère probable », la forme « sévère possible » et la forme « légère probable » (il n’existe pas de forme « légère possible »).
La notion de « probable » repose sur les critères suivants :
- mutation génétique indiquée par l’histoire familiale ou les tests
- ou présence des 3 sous-critères suivants :
- évidence claire d’un déclin de la mémoire et des capacités d’apprentissage et d’un autre domaine cognitif ;
- et déclin continu et graduel sans plateau prolongé ;
- et aucune évidence d’étiologie mixte.
En cas de forme sévère ne respectant pas les critères de « probable », la forme « sévère possible » est retenue.
1.1.1.4 Critères diagnostiques de la CIM-10 (1990)
Le diagnostic de maladie d’Alzheimer repose sur les 4 critères suivants :
- critères de démence ;
- et début insidieux et détérioration lentement progressive. Le début des troubles est habituellement difficile à déceler et l’entourage prend parfois brusquement conscience de la présence d’une détérioration. Le trouble peut sembler se stabiliser au cours de l’évolution ;
- et absence d’argument, d’après l’examen clinique et les investigations complémentaires en faveur d’une autre maladie somatique ou cérébrale (hypothyroïdie, hypercalcémie, carence en vitamine B12, carence en acide nicotinique, neuro-syphilis, hydrocéphalie à pression normale, hématome sous-dural) ;
- et début non brutal et absence, à un stade précoce de l’évolution de signes neurologiques d’une atteinte en foyer (hémiparésie, déficit sensoriel, déficit du champ visuel ou incoordination). Ces manifestations peuvent se surajouter secondairement.
Les critères diagnostiques de la CIM-11 (2019) évoquent un déclin « lent mais constant (de la mémoire) par rapport à un niveau antérieur de fonctionnement cognitif, avec une déficience dans d’autres domaines cognitifs (fonctions exécutives, attention, langage, cognition et jugement sociaux, vitesse psychomotrice, capacités visuo-perceptuelles ou visuo-spatiales) ». Selon la CIM-11, les tests génétiques positifs, les antécédents familiaux et le déclin cognitif graduel « suggèrent fortement » une démence due à la maladie d’Alzheimer.
1.1.1.5 Diagnostic de certitude anatomopathologique
La révision des critères NINCDS-ADRA (2007) a permis pour la première fois de retenir le diagnostic de maladie d’Alzheimer « définie » sur des critères cliniques et génétiques. A l’exception de ces critères, le diagnostic de certitude reste anatomopathologique [31]:
- détection de plaques séniles constituées de peptides amyloïdes bêta, et dépôt extracellulaire de peptides amyloïdes bêta solubles, produits par la voie amyloïdogène via le clivage d’APP (amyloid precursor protein) par les bêta- et gamma-secrétase (la voie non amyloïdogène et non pathogène faisant intervenir l’alpha-secrétase) ;
- et dégénérescence neurofibrillaire : accumulation intracellulaire de protéine Tau hyperphosphorylée, au niveau du locus cœruleuspuis s’étendant au niveau des cortex temporal, frontal et pariétal.
D’autres lésions anatomopathologiques peuvent être observées. Dans la moitié des cas, il existe à l’autopsie des signes d’autres démences associées : lésions vasculaires, angiopathie [32], sclérose hippocampique [33], corps de Lewy ou accumulation d’alpha-synucléine [34], inclusions immunoréactives de TDP-43 (Transactive response DNA Binding Protein43 kD[35,36]). On parle alors de démence mixte lorsque le diagnostic est évoqué [37].
1.1.2 Démence vasculaire
La démence vasculaire, secondaire à une maladie cérébrovasculaire ischémique ou hémorragique, est estimée à environ 15 % des démences [38]. L’apparition des déficits cognitifs est liée dans le temps à un ou plusieurs évènements vasculaires : avoir un accident vasculaire cérébral double le risque de démence, avec un risque proche de 50 % après 25 ans d’évolution [39].
La démence vasculaire inclut les démences par infarctus multiples, les démences par infarctus unique, les démences vasculaires ischémiques sous-corticales, les démences vasculaires hémorragiques, les démences secondaires à une hypoperfusion cérébrale chronique [40].
Le déclin cognitif concerne souvent la rapidité du traitement de l’information, l’attention complexe et les fonctions frontales. Ainsi, contrairement à la démence d’Alzheimer, les signes initiaux ne sont pas la perte de mémoire mais les troubles du jugement et de la prise de décision ou de planification. Ces troubles sont souvent associés à d’autres symptômes : troubles de la marche précoces (marche à petits pas), troubles de l’équilibre (instabilité, chutes), troubles du contrôle mictionnel, paralysie pseudo-bulbaire et « incontinence émotionnelle », modifications de la personnalité et de l’humeur, troubles moteurs [37].
Le premier score clinique utilisé pour différencier la démence vasculaire de la démence d’Alzheimer est le Hachinski Ischemic Score(score ≥ 7 pour la première, ≤ 4 pour la seconde), avec une sensibilité et une spécificité estimées à 89 % ; sa performance diagnostique est néanmoins médiocre pour distinguer les démences mixtes et les démences sous-corticales [41].
Selon le concept initial, les maladies vasculaires cérébrales entraînaient une démence lorsqu’il y avait de nombreux infarctus corticaux [42,43]. Le DSM-IV et la CIM-10 ont utilisé ce concept d’infarctus multiples pour définir la démence vasculaire [38,44].
Les progrès de l’imagerie cérébrale, de la génétique et de l’évolution du concept de démence ont conduit à l’individualisation de différents sous-types de démence vasculaire dans la CIM-10 : infarctus multiples (corticaux et/ou sous-corticaux), démence vasculaire sous-corticale, démence vasculaire à déclenchement aigu, types mixtes.
Plusieurs critères diagnostiques ont été proposés. En 1992 et 1993, les Alzheimer’s Disease Diagnostic and Treatment Centers(ADDTC) puis le NINDS-AIREN ont proposé des critères pour le diagnostic de la démence vasculaire [45,46]. Une des principales différences de ces critères était la notion d’au moins 2 accidents vasculaires cérébraux pour les critères ADDTC. Ces critères ont été comparés : en raison de leur grande spécificité, les critères NINDS-AIREN ont été préférés dans la plupart des études [38,47]. Ils définissent la démence vasculaire probable comme :
- une démence : déclin cognitif par rapport à un niveau antérieur, avec déclin mnésique et au moins deux autres domaines cognitifs, interférant avec les activités de la vie quotidienne ;
- et une maladie cérébrovasculaire : présence de signes focaux à l’examen neurologique et preuve par scanner ou IRM de lésion vasculaire cérébrale ;
- et une relation entre les deux affections, traduite par le début de la démence dans les 3 mois après un AVC ou une détérioration brusque des fonctions cognitives, ou une aggravation fluctuante ou par à-coups (en marche d’escalier).
Si l’imagerie est manquante, si la relation temporelle n’est pas claire, ou si le début est insidieux à évolution variable (plateau, amélioration), la démence vasculaire est dite possible.
A l’inverse, la démence vasculaire peut être certaine si les 3 éléments suivants sont réunis :
- critères d’une démence vasculaire probable ;
- et signes histopathologiques de maladie cérébrovasculaire ;
- et absence de dégénérescence neurofibrillaire, de plaques séniles et d’autres affections cliniques ou neuropathologiques pouvant être la cause de la démence.
En 2000, Erkinjuntti et al. ont proposé une modification de ces critères NINDS-AIREN pour la démence vasculaire sous-corticale [48]. En 2005, deux revues de littérature ont souligné l’importance de la démence vasculaire après un accident vasculaire cérébral [39,49]. En 2013, une revue française sur les concepts actuels de la démence vasculaire incluait non seulement les démences après infarctus corticaux et/ou sous-corticaux multiples, mais aussi celles associées aux infarctus uniques ou stratégiques, aux lésions de la substance blanche sans infarctus, aux hémorragies et à l’hypoperfusion [40]. Enfin, en 2014, la déclaration VASCOG a proposé de nouveaux critères [50].
1.1.3 Démence à corps de Lewy, maladie de Parkinson
1.1.3.1 Démence à corps de Lewy
La démence à corps de Lewy représente environ 4 à 7 % des démences [51,52]. Elle a été décrite en 1961 par Okazaki, Lipkin et Aronson [53]. Elle est plus fréquemment associée à une démence d’Alzheimer dans le cadre d’une démence mixte [37].
Son étiologie précise n’est pas connue, mais implique une accumulation de protéines alpha-synucléine dans les neurones (corps de Lewy). Sur une série autoptique de 120 cerveaux, une pathologie à corps de Lewy était présente chez 20 % des patients étiquetés avec une maladie d’Alzheimer [54]. Une dégénérescence neurofibrillaire et des plaques bêta-amyloïdes peuvent également être trouvées.
La plainte initiale concerne surtout les déficits d’attention et des fonctions exécutives. Ces troubles cognitifs s’accompagnent d’hallucinations visuelles (ou d’autres modalités sensorielles) et troubles du sommeil paradoxal. Des troubles dépressifs et idées délirantes peuvent être présents, ainsi qu’une dysautonomie (incontinence, hypotension orthostatique) [55,56]. L’apparition d’un syndrome parkinsonien dans l’année suivant les troubles cognitifs est caractéristique [17].
1.1.3.2 Démence au cours d’une maladie de Parkinson
Au cours de l’évolution de la maladie de Parkinson, une démence secondaire est possible par accumulation de corps de Lewy dans le cortex (comme dans la démence à corps de Lewy) ou par accumulation d’amas bêta-amyloïdes et de protéines tau (comme dans la démence d’Alzheimer) [37].
1.1.4 Démence mixte
Les démences mixtes sont caractérisées par la concomitance de plusieurs causes de démence [37]. Dans une étude autoptique, il a été montré que la moitié des personnes ayant une démence ont plusieurs causes associées : démence d’Alzheimer et démence vasculaire (38 %), démence d’Alzheimer et démence à corps de Lewy (12 %), association des 3 (2 %) [57]. L’association d’une démence vasculaire à une maladie d’Alzheimer dans le cadre d’une démence mixte est fréquente : environ 50 % des patients des patients âgés avec une démence d’Alzheimer ont des preuves anatomopathologiques d’infarctus silencieux [58]. Les mécanismes physiopathologiques sont intriqués avec ceux de la maladie d’Alzheimer [59,60].
1.1.5 Dégénérescence lobaire fronto-temporale
La dégénérescence lobaire fronto-temporale est un groupe de troubles neurodégénératifs représentant environ 10 % des démences (3 à 26 % selon les études) [61,62].
Plusieurs mécanismes peuvent être impliqués mais la (ou les) cause(s) de la maladie sont inconnues. Elle est associée à une pathologie de la protéine Tau, de la protéine TDP-43 (type A, B, C ou D), ou plus rarement des protéines FUS ou UPS [63]. Certaines formes ne s’accompagnent pas de lésion histologique spécifique [63–65].
La dégénérescence lobaire fronto-temporale est caractérisée par un déficit progressif du comportement (apathie, désinhibition, détérioration de la cognition sociale, comportements répétitifs, changements alimentaires) et/ou du langage (diminution de la parole, écholalie, aphasie progressive), associée à une détérioration intellectuelle. Contrairement à la maladie d’Alzheimer, la mémoire est classiquement préservée initialement, et les troubles débutent plus souvent entre 45 et 60 ans [37]. Plusieurs variantes syndromiques sont décrites, notamment BV(behavioural variant), SV-PPA(primary progressive aphasia – semantic variant) et NFV-PPA(primary progressive aphasia – non-fluent variant) [62]. D’autres types de démence peuvent être incluses dans la dégénérescence lobaire fronto-temporale selon leur mode de début ou l’anatomopathologie : maladie de Pick, dégénérescence cortico-basale, paralysie supra-nucléaire progressive (maladie de Steele-Richardson-Olszewski)[37].
1.1.6 Démence secondaire à l’abus de substances psychoactives
La démence peut être secondaire à l’abus de substances psychoactives. Il s’agit d’un diagnostic d’élimination ; pour le retenir, la quantité et la durée de consommation doivent être suffisantes pour entraîner la déficience cognitive, qui ne peut pas être mieux représentée par un autre type de démence.
1.1.6.1 Tout type de substances
Les principales substances à risque sont l’alcool, les benzodiazépines et apparentés (hypnotiques), les substances volatiles inhalées [66], les antiépileptiques [67]. D’autres psychotropes tels que les antidépresseurs et antipsychotiques ont été évoqués dans une étude cas/non-cas dans la base nationale de pharmacovigilance [68]. Les anticholinergiques ont aussi été associés à un sur-risque de maladie d’Alzheimer, et restent souvent prescrits dans cette population [69–71]. Une échelle du « fardeau cognitif des anticholinergiques » (Anticholinergic Cognitive Burden) a été publiée en 2008 [72]et est présentée en Annexe 2.
1.1.6.2 Cas particulier de l’alcool
Parmi ces substances à risque, l’alcool occupe une place particulière [73]. A partir de 1997, une consommation faible à modérée d’alcool a été proposée comme facteur de protection contre le développement de la démence [74–84]. Ces conclusions contrastent avec d’autres, estimant qu’une consommation faible à modérée d’alcool est un facteur de risqué[85–87]ou un facteur non associé [88–96].
La consommation excessive chronique d’alcool semble, elle, liée à un risque accru de démence. Elle peut également causer d’autres dommages cérébraux (neurotoxicité directe de l’éthanol, déficience en thiamine, syndrome de Wernicke-Korsakoff, encéphalopathie hépatique, démence vasculaire) et être associée à d’autres facteurs de risque de démence (tabagisme, moins de scolarité, dépression) [97–99].
Comme les études d’intervention ne sont pas éthiquement réalisables pour l’exposition à l’alcool, les meilleures données probantes proviennent d’études épidémiologiques.
Au début de 2013, cette littérature a donné lieu à une discussion sur l’introduction d’un diagnostic spécifique de démence liée à l’alcool [100]. Fin 2015, un document d’orientation publié par le National Institute for Health and Care Excellence(NICE) soulignait que les personnes âgées de 40 à 64 ans qui consomment régulièrement de l’alcool présentent un risque accru de démence [101,102]. En 2016 et 2018, une communication a été faite au sujet d’une étude analysant la base du Programme de Médicalisation des Systèmes d’Information (PMSI) entre 2008 et 2013, qui a établi un lien entre la démence précoce et les troubles liés à la consommation d’alcool [99,103].
1.1.7 Troubles cognitifs liés à d’autres pathologies
1.1.7.1 Hydrocéphalie à pression normale
L’hydrocéphalie à pression normale est une pathologie relativement fréquente, avec une prévalence estimée à environ 0,02 % pour les patients de plus de 40 ans et 1,4 % pour les patients de plus de 65 ans [104,105]. Les symptômes initiaux sont l’ataxie (marche à petits pas), les troubles urinaires et les troubles mnésiques (wobbly, weird and wet) [106]. Elle représente moins de 5 % des cas de démence [106].
1.1.7.2 Atrophies progressives focales
Les atrophies progressives focales réalisent des tableaux cognitivo-comportementaux, thymiques et moteurs d’évolution progressive, commençant généralement autour de 50 ans. Les principales atrophies progressives focales sont représentées par les atrophies fronto-temporales, les atrophies hémisphériques, les formes purement motrices focales progressives ou les atrophies corticales postérieures [64].
Cette dernière entité a été décrite par Benson en 1988 ; cette forme rare et récemment décrite a notamment été médiatisée par l’auteur britannique Terry Pratchett, qui en a souffert. Elle désigne un syndrome clinique dans lequel le traitement visuel d’ordre supérieur est perturbé en raison d’une maladie neurodégénérative. Les patients présentent une agnosie visuelle progressive et sévère (incapacité de reconnaître et d’identifier des objets ou des personnes familiers) et une apraxie (perte de la capacité d’exécuter ou d’exécuter des mouvements familiers qualifiés : alexie, agraphie spatiale, dysorthographie, acalculie, désorientation topographique, etc.) [17].
1.1.7.3 Maladie de Huntington
La maladie de Huntington est une maladie héréditaire rare, liée à la répétition du triplet CAG au niveau du gène de l’huntingtine (gène HTT) [107]. Les premiers symptômes se développent en général autour de 40 ans (parfois dès l’adolescence). Les troubles cognitifs s’installent progressivement avec un ralentissement de la pensée, des troubles de la mémoire et de l’attention jusqu’à une démence sous-corticale. Il s’y associe des signes moteurs (chorée, dystonie, troubles de la coordination, akinésie, troubles de l’élocution et de la déglutition, etc.) et des troubles du comportement (anxiété, dépression, irritabilité, apathie, désinhibition, etc.) [108,109]
1.1.7.4 Maladie de Creutzfeld-Jakob
Une cause rare mais rapidement évolutive de démence est la maladie de Creutzfeld-Jakob, décrite en 1920. Elle est caractérisée par des troubles mnésiques, de coordination et de comportement. Elle est secondaire au mauvais repliement d’une protéine (prion) qui provoque le dysfonctionnement d’autres protéines cérébrales. La maladie à prion la plus courante chez l’humain est la forme sporadique, dont la voie de propagation et le mécanisme de développement sont encore inconnus ou hypothétiques. En outre, on connaît les formes iatrogènes (transplantations de dure-mère, gonadotrophines, greffes de cornée), les formes génétiques et la forme variante [37,110].
Cette dernière (forme variante) est apparue en 1996, liée à l’ingestion de produits bovins infectés par l’encéphalopathie spongiforme bovine (maladie de la vache folle) ; moins de 200 cas ont été rapportés dans le monde, essentiellement au Royaume-Uni [111]. En raison de la résistance remarquable des prions aux procédures de stérilisation conventionnelles, une surveillance et une analyse minutieuses des facteurs de risque potentiels sont nécessaires.
1.1.7.5 Autres démences et diagnostics différentiels
Enfin, à côté des démences liées à la maladie de Parkinson, à la maladie de Huntington, au prion, ou à l’hydrocéphalie à pression normale, il existe d’autres formes de démences dues à d’autres maladies (et classées comme telles dans la CIM-10 et la CIM-11) :
- exposition à des métaux lourds ;
- virus de l’immunodéficience humaine (VIH) ;
- sclérose en plaques ;
- carence en pellagre ;
- trisomie 21.
Il existe des formes de démences liées à d’autres maladies : tumeurs bénignes (méningiomes), hématome sous-dural chronique, neurosyphilis, neuro-Lyme, neuro-Whipple, maladie de Behçet, neuro-sarcoïdose, dysnatrémie, hypercalcémie, dysthyroïdie, carence en vitamine B12 [112]. Enfin, il existe des pathologies plus rares : leucodystrophies, pathologies lysosomiales, mitochondriales, des ganglions basaux [113].
—
Références bibliographiques
[21] Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention, and care. The Lancet 2017;390:2673–734. doi:10.1016/S0140-6736(17)31363-6.
[22] Garre-Olmo J, Flaqué M, Gich J, Pulido TO, Turbau J, Vallmajo N, et al. A clinical registry of dementia based on the principle of epidemiological surveillance. BMC Neurol 2009;9:5. doi:10.1186/1471-2377-9-5.
[23] Garre-Olmo J. [Epidemiology of Alzheimer’s disease and other dementias]. Rev Neurol 2018;66:377–86.
[24] Harvey RJ, Skelton-Robinson M, Rossor MN. The prevalence and causes of dementia in people under the age of 65 years. J Neurol Neurosurg Psychiatry 2003;74:1206–9.
[25] Mercy L, Hodges JR, Dawson K, Barker RA, Brayne C. Incidence of early-onset dementias in Cambridgeshire, United Kingdom. Neurology 2008;71:1496–9. doi:10.1212/01.wnl.0000334277.16896.fa.
[26] Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med 2011;3:89ra57. doi:10.1126/scitranslmed.3002156.
[27] Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci 2014;8:113. doi:10.3389/fncel.2014.00113.
[28] McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939–44.
[29] Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007;6:734–46. doi:10.1016/S1474-4422(07)70178-3.
[30] Varma AR, Snowden JS, Lloyd JJ, Talbot PR, Mann DMA, Neary D. Evaluation of the NINCDS-ADRDA criteria in the differentiation of Alzheimer’s disease and frontotemporal dementia. J Neurol Neurosurg Psychiatry 1999;66:184–8. doi:10.1136/jnnp.66.2.184.
[31] Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2012;8:1–13. doi:10.1016/j.jalz.2011.10.007.
[32] Thal DR, Griffin WST, de Vos RAI, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol (Berl) 2008;115:599–609. doi:10.1007/s00401-008-0366-2.
[33] Amador-Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis. Handb Clin Neurol 2008;89:569–72. doi:10.1016/S0072-9752(07)01253-5.
[34] Leverenz JB, Fishel MA, Peskind ER, Montine TJ, Nochlin D, Steinbart E, et al. Lewy body pathology in familial Alzheimer disease: evidence for disease- and mutation-specific pathologic phenotype. Arch Neurol 2006;63:370–6. doi:10.1001/archneur.63.3.370.
[35] Wilson AC, Dugger BN, Dickson DW, Wang D-S. TDP-43 in aging and Alzheimer’s disease – a review. Int J Clin Exp Pathol 2011;4:147–55.
[36] James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer’s-type dementia. Brain J Neurol 2016. doi:10.1093/brain/aww224.
[37] 2018 Alzheimer’s disease facts and figures. Alzheimers Dement J Alzheimers Assoc 2018;14:367–429. doi:10.1016/j.jalz.2018.02.001.
[38] O’Brien JT, Thomas A. Vascular dementia. Lancet Lond Engl 2015;386:1698–706. doi:10.1016/S0140-6736(15)00463-8.
[39] Leys D, Hénon H, Mackowiak-Cordoliani M-A, Pasquier F. Poststroke dementia. Lancet Neurol 2005;4:752–9. doi:10.1016/S1474-4422(05)70221-0.
[40] Benisty S. Démences vasculaires : concepts actuels. Gériatrie Psychol Neuropsychiatr Vieil 2013;11:171–80. doi:10.1684/pnv.2013.0410.
[41] Moroney JT, Bagiella E, Desmond DW, Hachinski VC, Mölsä PK, Gustafson L, et al. Meta-analysis of the Hachinski Ischemic Score in pathologically verified dementias. Neurology 1997;49:1096–105.
[42] Hachinski VC, Lassen NA, Marshall J. Multi-infarct dementia. A cause of mental deterioration in the elderly. Lancet Lond Engl 1974;2:207–10.
[43] McKay E, Counts SE. Multi-Infarct Dementia: A Historical Perspective. Dement Geriatr Cogn Disord Extra 2017;7:160–71. doi:10.1159/000470836.
[44] Perneczky R, Tene O, Attems J, Giannakopoulos P, Ikram MA, Federico A, et al. Is the time ripe for new diagnostic criteria of cognitive impairment due to cerebrovascular disease? Consensus report of the International Congress on Vascular Dementia working group. BMC Med 2016;14:162. doi:10.1186/s12916-016-0719-y.
[45] Chui HC, Victoroff JI, Margolin D, Jagust W, Shankle R, Katzman R. Criteria for the diagnosis of ischemic vascular dementia proposed by the State of California Alzheimer’s Disease Diagnostic and Treatment Centers. Neurology 1992;42:473–473. doi:10.1212/WNL.42.3.473.
[46] Román GC, Tatemichi TK, Erkinjuntti T, Cummings JL, Masdeu JC, Garcia JH, et al. Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 1993;43:250–60.
[47] Pohjasvaara T, Mäntylä R, Ylikoski R, Kaste M, Erkinjuntti T. Comparison of different clinical criteria (DSM-III, ADDTC, ICD-10, NINDS-AIREN, DSM-IV) for the diagnosis of vascular dementia. National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences. Stroke 2000;31:2952–7.
[48] Erkinjuntti T, Inzitari D, Pantoni L, Wallin A, Scheltens P, Rockwood K, et al. Research criteria for subcortical vascular dementia in clinical trials. J Neural Transm Suppl 2000;59:23–30.
[49] Hénon H, Pasquier F, Leys D. Poststroke dementia. Cerebrovasc Dis Basel Switz 2006;22:61–70. doi:10.1159/000092923.
[50] Sachdev P, Kalaria R, O’Brien J, Skoog I, Alladi S, Black SE, et al. Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis Assoc Disord 2014;28:206–18. doi:10.1097/WAD.0000000000000034.
[51] Walker Z, Possin KL, Boeve BF, Aarsland D. Lewy body dementias. Lancet Lond Engl 2015;386:1683–97. doi:10.1016/S0140-6736(15)00462-6.
[52] Hogan DB, Fiest KM, Roberts JI, Maxwell CJ, Dykeman J, Pringsheim T, et al. The Prevalence and Incidence of Dementia with Lewy Bodies: a Systematic Review. Can J Neurol Sci J Can Sci Neurol 2016;43 Suppl 1:S83-95. doi:10.1017/cjn.2016.2.
[53] Okazaki H, Lipkin LE, Aronson SM. Diffuse intracytoplasmic ganglionic inclusions (Lewy type) associated with progressive dementia and quadriparesis in flexion. J Neuropathol Exp Neurol 1961;20:237–44.
[54] De Reuck J, Deramecourt V, Cordonnier C, Pasquier F, Leys D, Maurage C-A, et al. The incidence of post-mortem neurodegenerative and cerebrovascular pathology in mixed dementia. J Neurol Sci 2016;366:164–6. doi:10.1016/j.jns.2016.05.021.
[55] McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47:1113–24.
[56] McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017;89:88–100. doi:10.1212/WNL.0000000000004058.
[57] Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007;69:2197–204. doi:10.1212/01.wnl.0000271090.28148.24.
[58] Fernando MS, Ince PG. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci 2004;226:13–7. doi:10.1016/j.jns.2004.09.004.
[59] Kalaria RN. Cerebrovascular disease and mechanisms of cognitive impairment: evidence from clinicopathological studies in humans. Stroke 2012;43:2526–34. doi:10.1161/STROKEAHA.112.655803.
[60] Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, et al. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2015;11:710–7. doi:10.1016/j.jalz.2014.10.008.
[61] Vieira RT, Caixeta L, Machado S, Silva AC, Nardi AE, Arias-Carrión O, et al. Epidemiology of early-onset dementia: a review of the literature. Clin Pract Epidemiol Ment Health CP EMH 2013;9:88–95. doi:10.2174/1745017901309010088.
[62] Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet Lond Engl 2015;386:1672–82. doi:10.1016/S0140-6736(15)00461-4.
[63] Li Y-Q, Tan M-S, Yu J-T, Tan L. Frontotemporal Lobar Degeneration: Mechanisms and Therapeutic Strategies. Mol Neurobiol 2016;53:6091–105. doi:10.1007/s12035-015-9507-5.
[64] Ghika J, Joray S, Brioschi A, Frackowiak RSJ. Atrophies focales. EMC – Neurol 2010;7:1–14.
[65] Seltman RE, Matthews BR. Frontotemporal lobar degeneration: epidemiology, pathology, diagnosis and management. CNS Drugs 2012;26:841–70. doi:10.2165/11640070-000000000-00000.
[66] Howard MO, Bowen SE, Garland EL, Perron BE, Vaughn MG. Inhalant Use and Inhalant Use Disorders in the United States. Addict Sci Clin Pract 2011;6:18–31.
[67] Bottino CMC, de Pádua AC, Smid J, Areza-Fegyveres R, Novaretti T, Bahia VS, et al. Differential diagnosis between dementia and psychiatric disorders: Diagnostic criteria and supplementary exams. Recommendations of the Scientific Department of Cognitive Neurology and Aging of the Brazilian Academy of Neurology. Dement Neuropsychol 2011;5:288–96. doi:10.1590/S1980-57642011DN05040006.
[68] Favrelière S, Lafay-Chebassier C, Alkhidir F, Merlet I, Pérault Pochat M-C. [Drug-induced dementia: a case/non-case study in the French Pharmacovigilance database]. Therapie 2007;62:507–11. doi:10.2515/therapie:2007070.
[69] Gray SL, Anderson ML, Dublin S, Hanlon JT, Hubbard R, Walker R, et al. Cumulative use of strong anticholinergics and incident dementia: a prospective cohort study. JAMA Intern Med 2015;175:401–7. doi:10.1001/jamainternmed.2014.7663.
[70] Montastruc F, Gardette V, Cantet C, Piau A, Lapeyre-Mestre M, Vellas B, et al. Potentially inappropriate medication use among patients with Alzheimer disease in the REAL.FR cohort: be aware of atropinic and benzodiazepine drugs! Eur J Clin Pharmacol 2013;69:1589–97. doi:10.1007/s00228-013-1506-8.
[71] Montastruc F, Rouanet S, Gardette V, Rousseau V, Bagheri H, Montastruc J-L. Atropinic burden of prescriptions forms in patients with Alzheimer disease: a cross-sectional study in a French PharmacoVigilance Database. Eur J Clin Pharmacol 2015;71:891–5. doi:10.1007/s00228-015-1869-0.
[72] Boustani M, Campbell N, Munger S, Maidment I, Fox C. Impact of anticholinergics on the aging brain: a review and practical application. Aging Health 2008;4:311–20. doi:10.2217/1745509X.4.3.311.
[73] Piazza-Gardner AK, Gaffud TJB, Barry AE. The impact of alcohol on Alzheimer’s disease: A systematic review. Aging Ment Health 2013;17:133–46. doi:10.1080/13607863.2012.742488.
[74] Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, et al. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997;153:185–92.
[75] Huang W, Qiu C, Winblad B, Fratiglioni L. Alcohol consumption and incidence of dementia in a community sample aged 75 years and older. J Clin Epidemiol 2002;55:959–64.
[76] Lindsay J, Laurin D, Verreault R, Hébert R, Helliwell B, Hill GB, et al. Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol 2002;156:445–53.
[77] Bachman DL, Green RC, Benke KS, Cupples LA, Farrer LA, MIRAGE Study Group. Comparison of Alzheimer’s disease risk factors in white and African American families. Neurology 2003;60:1372–4.
[78] Deng J, Zhou DHD, Li J, Wang YJ, Gao C, Chen M. A 2-year follow-up study of alcohol consumption and risk of dementia. Clin Neurol Neurosurg 2006;108:378–83. doi:10.1016/j.clineuro.2005.06.005.
[79] Ogunniyi A, Hall KS, Gureje O, Baiyewu O, Gao S, Unverzagt FW, et al. Risk factors for incident Alzheimer’s disease in African Americans and Yoruba. Metab Brain Dis 2006;21:235–40. doi:10.1007/s11011-006-9017-2.
[80] Peters R, Peters J, Warner J, Beckett N, Bulpitt C. Alcohol, dementia and cognitive decline in the elderly: a systematic review. Age Ageing 2008;37:505–12. doi:10.1093/ageing/afn095.
[81] Anstey KJ, Mack HA, Cherbuin N. Alcohol consumption as a risk factor for dementia and cognitive decline: meta-analysis of prospective studies. Am J Geriatr Psychiatry 2009;17:542–555.
[82] García AM, Ramón-Bou N, Porta M. Isolated and joint effects of tobacco and alcohol consumption on risk of Alzheimer’s disease. J Alzheimers Dis JAD 2010;20:577–86. doi:10.3233/JAD-2010-1399.
[83] Neafsey EJ, Collins MA. Moderate alcohol consumption and cognitive risk. Neuropsychiatr Dis Treat 2011;7:465–84. doi:10.2147/NDT.S23159.
[84] Weyerer S, Schäufele M, Wiese B, Maier W, Tebarth F, van den Bussche H, et al. Current alcohol consumption and its relationship to incident dementia: results from a 3-year follow-up study among primary care attenders aged 75 years and older. Age Ageing 2011;40:456–63. doi:10.1093/ageing/afr007.
[85] Fratiglioni L, Ahlbom A, Viitanen M, Winblad B. Risk factors for late-onset Alzheimer’s disease: a population-based, case-control study. Ann Neurol 1993;33:258–66. doi:10.1002/ana.410330306.
[86] Harwood DG, Barker WW, Loewenstein DA, Ownby RL, St George-Hyslop P, Mullan M, et al. A cross-ethnic analysis of risk factors for AD in white Hispanics and white non-Hispanics. Neurology 1999;52:551–6.
[87] Harwood DG, Kalechstein A, Barker WW, Strauman S, St George-Hyslop P, Iglesias C, et al. The effect of alcohol and tobacco consumption, and apolipoprotein E genotype, on the age of onset in Alzheimer’s disease. Int J Geriatr Psychiatry 2010;25:511–8. doi:10.1002/gps.2372.
[88] Graves AB, van Duijn CM, Chandra V, Fratiglioni L, Heyman A, Jorm AF, et al. Alcohol and tobacco consumption as risk factors for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. Int J Epidemiol 1991;20 Suppl 2:S48-57.
[89] Hebert LE, Scherr PA, Beckett LA, Funkenstein HH, Albert MS, Chown MJ, et al. Relation of smoking and alcohol consumption to incident Alzheimer’s disease. Am J Epidemiol 1992;135:347–55.
[90] Rosen J, Colantonio A, Becker JT, Lopez OL, DeKosky ST, Moss HB. Effects of a history of heavy alcohol consumption on Alzheimer’s disease. Br J Psychiatry J Ment Sci 1993;163:358–63.
[91] Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, et al. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology 1995;45:1161–8.
[92] Tsolaki M, Fountoulakis K, Chantzi E, Kazis A. Risk factors for clinically diagnosed Alzheimer’s disease: a case-control study of a Greek population. Int Psychogeriatr 1997;9:327–41.
[93] Broe GA, Creasey H, Jorm AF, Bennett HP, Casey B, Waite LM, et al. Health habits and risk of cognitive impairment and dementia in old age: a prospective study on the effects of exercise, smoking and alcohol consumption. Aust N Z J Public Health 1998;22:621–3.
[94] Tyas SL, Koval JJ, Pederson LL. Does an interaction between smoking and drinking influence the risk of Alzheimer’s disease? Results from three Canadian data sets. Stat Med 2000;19:1685–96.
[95] Ruitenberg A, van Swieten JC, Witteman JCM, Mehta KM, van Duijn CM, Hofman A, et al. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet Lond Engl 2002;359:281–6. doi:10.1016/S0140-6736(02)07493-7.
[96] Luchsinger JA, Tang M-X, Siddiqui M, Shea S, Mayeux R. Alcohol intake and risk of dementia. J Am Geriatr Soc 2004;52:540–6. doi:10.1111/j.1532-5415.2004.52159.x.
[97] Kim JW, Lee DY, Lee BC, Jung MH, Kim H, Choi YS, et al. Alcohol and Cognition in the Elderly: A Review. Psychiatry Investig 2012;9:8–16. doi:10.4306/pi.2012.9.1.8.
[98] Kuźma E, Llewellyn DJ, Langa KM, Wallace RB, Lang IA. History of Alcohol Use Disorders and Risk of Severe Cognitive Impairment: A 19-Year Prospective Cohort Study. Am J Geriatr Psychiatry 2014;22:1047–54. doi:10.1016/j.jagp.2014.06.001.
[99] Schwarzinger M, Pollock BG, Hasan OSM, Dufouil C, Rehm J, Baillot S, et al. Contribution of alcohol use disorders to the burden of dementia in France 2008–13: a nationwide retrospective cohort study. Lancet Public Health 2018;3:e124–32. doi:10.1016/S2468-2667(18)30022-7.
[100] Ridley NJ, Draper B, Withall A. Alcohol-related dementia: an update of the evidence. Alzheimers Res Ther 2013;5:3. doi:10.1186/alzrt157.
[101] Dementia, disability and frailty in later life – mid-life approaches to delay or prevent onset | Guidance and guidelines | NICE n.d. https://www.nice.org.uk/guidance/ng16 (accessed July 22, 2018).
[102] Warning about middle-aged drinking and dementia. NhsUk 2015. https://www.nhs.uk/news/neurology/warning-about-middle-aged-drinking-and-dementia/ (accessed July 20, 2018).
[103] Schwarzinger M, Thiebaut SP, Rehm J. THE NEGLECTED ROLE OF ALCOHOL USE DISORDERS IN EARLY-ONSET DEMENTIA: A NATIONWIDE STUDY FROM FRANCE. Alzheimers Dement J Alzheimers Assoc 2016;12:P194–5. doi:10.1016/j.jalz.2016.06.338.
[104] Brean A, Eide PK. Prevalence of probable idiopathic normal pressure hydrocephalus in a Norwegian population. Acta Neurol Scand 2008;118:48–53. doi:10.1111/j.1600-0404.2007.00982.x.
[105] Tanaka N, Yamaguchi S, Ishikawa H, Ishii H, Meguro K. Prevalence of Possible Idiopathic Normal-Pressure Hydrocephalus in Japan: The Osaki-Tajiri Project. Neuroepidemiology 2009;32:171–5. doi:10.1159/000186501.
[106] Rosseau G. Normal pressure hydrocephalus. Dis–Mon DM 2011;57:615–24. doi:10.1016/j.disamonth.2011.08.023.
[107] Nance MA. Genetics of Huntington disease. Handb Clin Neurol 2017;144:3–14. doi:10.1016/B978-0-12-801893-4.00001-8.
[108] Youssov K, Bachoud-Lévi A-C. Maladie de Huntington : aspects diagnostiques actuels et applications pratiques. EMC – Neurol 2008;5:1–13. doi:10.1016/S0246-0378(07)27525-6.
[109] Ghosh R, Tabrizi SJ. Clinical Features of Huntington’s Disease. Adv Exp Med Biol 2018;1049:1–28. doi:10.1007/978-3-319-71779-1_1.
[110] Kittner C, Heinemann U, Zerr I. [Risk factors for sporadic Creutzfeldt-Jakob disease]. Dtsch Med Wochenschr 1946 2009;134:1429–35. doi:10.1055/s-0029-1225299.
[111] University of Edinburgh. National CJD Research & Surveillance Unit 2017. http://www.cjd.ed.ac.uk/ (accessed September 16, 2018).
[112] Daher O, Nguyen S, Smith C, Bülab C, Démonet JF. Prise en charge et prevention des pathologies démentielles. Rev Médicale Suisse 2016;12:799–802.
[113] Ferrari C, Nacmias B, Sorbi S. The diagnosis of dementias: a practical tool not to miss rare causes. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol 2018;39:615–27. doi:10.1007/s10072-017-3206-0.
Partager :

